Data Requirements
A number of ancillary measurements are required to calculate in situ fugacity from the observed mole fraction of CO2 in dry air. Calculation of fugacities in the atmosphere is simpler since the dried air is directly injected into the analyzer (see Methods). Determination of the CO2 fugacity in surface seawater is not as straightforward. Firstly, the gas concentration is not measured in situ, since the water must be pumped up from the surface to the analysis site. Secondly, the gas concentration is not determined in the aqueous phase, but is determined in the vapor phase of the equilibrator. And thirdly, the measured quantity is in dried air rather than in the moist conditions of the seawater-air interface. Determining the in situ fugacity of CO2 in surface seawater requires eight pieces of information:
- concentration of the gas in the dried vapor phase from the seawater equilibrator
- in situ surface seawater temperature
- temperature of seawater in the equilibrator
- sea surface salinity
- atmospheric pressure
- relative humidity
- vapor pressure of seawater
- the functional relationship between gas solubility and temperature.
Some simplifying assumptions are generally made so that only three or four of these quantities are usually determined at sea. Each of the eight data requirements is discussed in more detail below.
(1.) The concentration of CO2 is measured in vapor drawn from the equilibrator. The vapor is dried by passing through a chemical trap and is then injected into an analyzer. The gas concentration is evaluated by comparison of the instrument response for an unknown concentration to the response for known concentrations of calibrated gas standards. Concentrations for CO2 gas standards are commonly expressed as mole fractions (the ratio of moles of gas of interest to the total moles of gas) in the dimensionless units of parts per million (ppm).
(2.) and (3.) In situ surface seawater temperature tss and the temperature of seawater in the equilibrator teq are measured directly. The analysis temperature, i.e., the temperature of seawater in the equilibrator teq, is generally higher than the in situ seawater temperature tss by about 0.5-1.0 °C. At least three reasons can account for the warming of water in the equilibrator: (i) Frictional warming occurs as the water travels from the seawater intake port to the equilibrator. The extent of this warming will depend on the distance traveled, flow rate, and on the inside diameter of the pipe, as well as on the type of pump used. (ii) Conductive warming may occur if a thermal gradient exists between the water traveling in the pipe and the pipe exterior. This effect is expected to be larger in cases where the pipe line runs mainly inside the ship. (iii) Warming resultant from insolation will be a factor in cases where the pipe runs outside the ship.
Gas solubiliy generally decreases with increasing temperature, as will be discussed later in more detail. If the seawater in the equilibrator is warmer than the in situ seawater, the apparent gas fugacity in the equilibrator will be higher than the fugacity in situ. The difference between the in situ gas fugacity and that measured in the equilibrator is ~4% per °C for CO2.
(4.) Salinity is either measured directly or assumed to be a constant value. Salinity is used for (7.) the evaluation of vapor pressure of seawater and (8.) the temperature and salinity dependence of gas solubility. For evaluation of the vapor pressure of seawater, the error resultant if salinity is ignored altogether is ~2%, and even smaller (~0.05%) if salinity is in error by 1 salinity unit. The error in neglecting salinity when calculating gas solubility is <0.1% for CO2.
(5.) Atmospheric pressure is either measured directly or assumed to be one atmosphere. If the atmospheric pressure at which the measurements had been made were 950 mbar instead of 1000 mbar, the fugacity of CO2 [f(CO2)] would be 5% lower. Only rarely does the atmospheric pressure deviate by this amount, and so more commonly the error in f(CO2) of assuming one atmosphere barometric pressure is smaller, perhaps 1-2%.
(6.) The relative humidity at the air-sea interface is assumed to be 100%, i.e., saturated in water vapor at the temperature of measurement. The gas concentration measured in dry air is adjusted to reflect the moist in situ condition. This assumption is a reasonable one since the vapor immediately above the sea surface is not likely to be far from saturated with water, but the error would be small in any event. If, for example, the humidity were 90% instead of 100%, the error in the in situ fugacity would be 0.3% for a mole fraction of 350 ppm gas in dry air.
(7.) The vapor pressure of seawater is generally calculated from the temperature and salinity data. One method of calculation is given by Weiss and Price (1980) in which one equation is used to assess the saturation vapor pressure over seawater. They combined the vapor pressure algorithm for pure water of Goff and Gratch (1946) with equations for vapor pressure lowering by sea salt given by Robinson (1954) and fit a polynomial in temperature and salinity over the range 273 to 313K and 0 to 40 ppt salinity:
ln psw = 24.4543-67.4509(100/T)-4.8489 ln (T/100)-0.000544S (eq. 1)
where:
psw is the vapor pressure of seawater
T is the temperature in Kelvin
S is the salinity in ppt
For natural seawater systems, the salinity correction is a small one. If, for example, salinity were assumed to be 0 instead of 33 psu (at 25 °C), the error in vapor pressure would be 1.8%, giving rise to a 0.06% error in calculating in situ gas fugacity for a measured mole fraction of 350 ppm.
(8.) The dependence of gas solubility on temperature is generally determined experimentally from laboratory studies. A functional relationship between gas solubility and temperature is established empirically from the data. As long as there is no chemical reaction between the solute gas and the solvent, the relationship can be used to infer gas solubility at a temperature other than the measured temperature. Since gas concentrations at sea are measured in equilibrator vapor at temperature teq, and teq is higher than in situ tss, the functional dependence of gas solubility on temperature might be used to calculate the gas solubility at the in situ temperature tss.
However, at normal seawater pH (~8), CO2 can react as follows:
CO2(g) + H2O <=> H2CO3 <=> HCO3- + H+ <=> CO32- + H+ (eq. 2)
For this reacting system a temperature change also induces changes in the chemical equilibria. The solubility of CO2 measured at normal seawater pH would include the effects of both solubility and reactivity of CO2, and would be too high. In order to determine only the solubility of CO2 gas, the experimental work on CO2 solubilities has been done with acidified solutions of seawater (pH ~2) to suppress ionization of CO2 according to equation 2 above. In order to determine a functional relationship between temperature and solubility of CO2 in a real seawater system, the reactivity of carbon dioxide in seawater must also be taken into account.
Weiss et al. (1982) evaluated the temperature dependence of f(CO2) in a gas phase equilibrated with seawater by calculating f(CO2) from equations describing carbonate system equilibria. For a given combination of salinity S, total titration alkalinity At, and dissolved inorganic carbon (DIC), the fugacity of CO2 was calculated over a temperature range of S, At, and DIC values. These data may be thought of as comprising a family of temperature vs. f(CO2) curves where each curve represents the fugacity dependence on temperature under slightly different chemical (S, At, and DIC) circumstances. The ideal situation, however, is one in which the temperature dependence of CO2 fugacity can be described independently of the chemical state. By considering the family of curves temperature vs. ln f(CO2), Weiss et al. (1982) fitted the natural logarithm of f(CO2) to a power series in temperature. Those expressions were differentiated, and again fitted to the following equation:
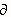 ln f(CO2)/t=0.03107-2.785 · 10-4 t-1.839 · 10-3 ln f(CO2) (eq. 3)
ln f(CO2)/t=0.03107-2.785 · 10-4 t-1.839 · 10-3 ln f(CO2) (eq. 3)
This equation indicates that, for a given f(CO2) and temperature, the increase in f(CO2) with temperature [i.e., the slope of the t vs. ln f(CO2) curve] for all these curves can be described by a power series in temperature. In this way, the temperature dependence of f(CO2) can be described without reference to the chemical state of the system. For every °C change in temperature, the f(CO2) changes by ~4%.